


Mô tả
Việc 21 CFR Part 820 chuyển sang QMSR và viện dẫn ISO 13485:2016 (hiệu lực 02/02/2026) khiến nhiều nhà sản xuất thiết bị y tế phải xây mới hoặc nâng cấp toàn bộ hệ thống quản lý chất lượng. Với đội ngũ chuyên gia ISO, tuân thủ và pháp lý, chúng tôi cung cấp dịch vụ tư vấn – đào tạo – hỗ trợ tuân thủ QMSR/ISO 13485 trọn gói, giúp doanh nghiệp Việt Nam và FDI tự tin tiếp cận thị trường Hoa Kỳ.
Vì sao chọn dịch vụ tư vấn QMSR của chúng tôi?
- Một đầu mối – hệ sinh thái tích hợp: ISC Global (tư vấn – đào tạo), STC VN / Staunchly Vietnam (triển khai) và Duc Luong Services (đối tác đại diện) phối hợp đồng bộ.
- Chuyên gia “lai” kỹ thuật + pháp lý: Hiểu sâu cả ISO 13485 lẫn các quy định FDA bổ sung (Part 803, 806, 821, 830), giúp doanh nghiệp tránh hiểu lầm “có ISO 13485 là đủ”.
- Song ngữ Việt – Anh: Thuận lợi cho doanh nghiệp FDI, làm việc với tổ chức chứng nhận quốc tế và chuẩn bị hồ sơ FDA.
- Thực chiến, cập nhật: Bám sát quy trình thanh tra mới (Compliance Program 7382.850) và bối cảnh chuyển đổi QSR → QMSR.
Lộ trình triển khai & chuyển đổi sang QMSR (8 bước)
Bước 1 – Phân tích khoảng cách (Gap Analysis) QSR → QMSR / ISO 13485
Đối chiếu hệ thống hiện tại với ISO 13485:2016 và các yêu cầu FDA bổ sung; lập ma trận đối chiếu (mapping) và kế hoạch khắc phục.
Bước 2 – Hoạch định hệ thống QMS
Thiết kế kiến trúc tài liệu, phân công trách nhiệm, xác định “applicable regulatory requirements” áp dụng cho từng dòng sản phẩm.
Bước 3 – Xây dựng & cập nhật tài liệu
Sổ tay chất lượng, quy trình theo ISO 13485 (kiểm soát thiết kế, mua hàng, sản xuất, validation, CAPA…) tích hợp yêu cầu FDA tại §820.10, §820.35, §820.45.
Bước 4 – Quản lý rủi ro (ISO 14971) & kiểm soát thiết kế
Tích hợp quản lý rủi ro xuyên suốt QMS; thiết lập hồ sơ thiết kế & phát triển (Design History File) đáp ứng Điều 7 ISO 13485.
Bước 5 – Đào tạo toàn diện
Đào tạo nhận thức QMSR/ISO 13485, đào tạo chuyên sâu theo vị trí và đào tạo đánh giá viên nội bộ (chi tiết bên dưới).
Bước 6 – Áp dụng, validation & vận hành thử
Hỗ trợ triển khai thực tế, xác nhận giá trị sử dụng quy trình/phần mềm, hoàn thiện hồ sơ và truy xuất nguồn gốc.
Bước 7 – Đánh giá nội bộ & CAPA
Tổ chức đánh giá nội bộ, mô phỏng thanh tra, xử lý điểm chưa phù hợp trước đánh giá/thanh tra chính thức.
Bước 8 – Hỗ trợ chứng nhận ISO 13485 & sẵn sàng thanh tra FDA / MDSAP
Kết nối tổ chức chứng nhận ISO 13485, hỗ trợ chuẩn bị hồ sơ và sẵn sàng cho thanh tra theo Compliance Program 7382.850; tư vấn lộ trình MDSAP nếu phù hợp.
Các gói dịch vụ
- Gói Chuyển đổi QSR → QMSR: Dành cho doanh nghiệp đã có hệ thống QSR cần cập nhật theo QMSR.
- Gói Xây dựng mới QMS theo ISO 13485 + QMSR: Từ con số 0 đến sẵn sàng chứng nhận/thanh tra.
- Gói Đào tạo QMSR/ISO 13485.
- Gói Đánh giá nội bộ & Mock FDA Inspection (tiền thanh tra).
- Gói Hỗ trợ MDSAP & mở rộng thị trường quốc tế.
Các khóa đào tạo tiêu biểu
- Nhận thức QMSR & ISO 13485:2016: cập nhật thay đổi 2026, điểm FDA bổ sung.
- Kiểm soát thiết kế & Quản lý rủi ro (ISO 14971).
- CAPA, kiểm soát sản phẩm không phù hợp, xử lý khiếu nại & MDR (Part 803).
- UDI (Part 830), theo dõi thiết bị (Part 821), khắc phục & thu hồi (Part 806).
- Đào tạo đánh giá viên nội bộ ISO 13485 / QMSR.
Hình thức linh hoạt: in-house, trực tuyến hoặc kết hợp, có kiểm tra và chứng nhận hoàn thành.
Những sai lầm thường gặp khi tự triển khai
- Cho rằng “có ISO 13485 là tuân thủ đủ QMSR” — bỏ sót yêu cầu FDA bổ sung.
- Quản lý rủi ro hình thức, không tích hợp xuyên suốt QMS.
- Hồ sơ thiết kế (DHF), validation thiếu bằng chứng.
- Hồ sơ khiếu nại/MDR không đáp ứng nội dung §820.35 và Part 803.
- Không mô phỏng thanh tra trước khi FDA đến.
Câu hỏi thường gặp (FAQ)
1. Chi phí và thời gian tư vấn QMSR là bao nhiêu? Phụ thuộc dòng sản phẩm, quy mô và hiện trạng. Vui lòng liên hệ để được khảo sát và báo giá minh bạch.
2. Chúng tôi chưa có gì, mất bao lâu để sẵn sàng? Tùy độ phức tạp thiết bị, thường 6–18 tháng cho hệ thống mới. Tư vấn bài bản giúp rút ngắn đáng kể.
3. Dịch vụ có hỗ trợ doanh nghiệp FDI và làm việc bằng tiếng Anh không? Có. Chúng tôi làm việc song ngữ Việt – Anh và phối hợp với tổ chức chứng nhận quốc tế.
4. Sau chứng nhận có hỗ trợ duy trì không? Có. Chúng tôi hỗ trợ đào tạo định kỳ, đánh giá nội bộ và chuẩn bị các kỳ đánh giá giám sát/thanh tra.
Liên hệ để được tư vấn cho doanh nghiệp
Hãy để chúng tôi đồng hành cùng bạn chinh phục thị trường thiết bị y tế Hoa Kỳ — tuân thủ đúng, sẵn sàng thanh tra, bền vững.
📞 Hotline: +84 933 096 426 – +84 868 591 260 ✉️ Email: info@iscglobal.asia | van.pham@iscglobal.asia 🌐 Website: iscglobal.asia | iscglobal.edu.vn
Đối tác đại diện tại Việt Nam – Duc Luong Services 📞 Hotline: +84 933 096 426 – +84 868 591 260 ✉️ Email: ducluongservices@gmail.com 🌐 Website: ducluongservices.com
STC VN Co., Ltd. (Staunchly Vietnam) 📞 Hotline: +84 933 096 426 – +84 868 591 260 ✉️ Email: info@staunchlyservices.com.vn 🌐 Website: staunchlyservices.com.vn
Khám Phá Thế Giới MedTech: Nhập Môn Xác Nhận Giá Trị Sử Dụng Quy Trình (Process Validation)
1. Tại Sao Quy Trình Lại Quan Trọng Đến Vậy?
Chào mừng bạn đến với hành trình chinh phục chất lượng trong ngành công nghiệp thiết bị y tế (MedTech). Đây là một lĩnh vực đặc thù, nơi mỗi quyết định kỹ thuật đều mang sức nặng của một lời cam kết sinh tử. Trong sản xuất thông thường, một sản phẩm lỗi có thể chỉ là tổn thất về kinh tế; nhưng trong y tế, một sai sót dù là nhỏ nhất cũng có thể tước đi cơ hội sống của một con người.
Làm thế nào để chúng ta đảm bảo hàng triệu sản phẩm xuất xưởng đều an toàn tuyệt đối, ngay cả khi chúng ta không thể “mổ xẻ” từng sản phẩm để kiểm tra? Câu trả lời nằm ở nghệ thuật và khoa học của Xác nhận giá trị sử dụng quy trình (Process Validation).
Theo định nghĩa cốt lõi từ FDA:
“Process Validation (Xác nhận giá trị sử dụng quy trình) nghĩa là thiết lập bằng bằng chứng khách quan rằng một quy trình tạo ra kết quả hoặc sản phẩm đáp ứng các thông số kỹ thuật đã định trước một cách nhất quán.”
Nhiều người mới bắt đầu thường thắc mắc: “Tại sao không chỉ kiểm tra sản phẩm sau khi làm xong cho đơn giản?”. Thực tế, có những thứ không thể nhìn thấy bằng mắt thường, và có những quy trình mà việc kiểm tra sẽ phá hủy chính sản phẩm đó. Hãy cùng tôi tìm hiểu “Quy tắc vàng” để biết khi nào chúng ta buộc phải Validation.
2. Quy Tắc Vàng: Khi Nào Bắt Buộc Phải Validation?
Để quản lý chất lượng, chúng ta có hai công cụ chính: Verification (Kiểm tra thành phẩm) và Validation (Xác nhận quy trình).
- Verification: Áp dụng khi bạn có thể kiểm tra 100% đầu ra mà không làm hỏng sản phẩm (ví dụ: đo kích thước của một chiếc kim tiêm).
- Validation: Áp dụng khi kết quả của quy trình không thể kiểm tra đầy đủ bằng các phép đo sau đó, hoặc các sai sót chỉ lộ ra khi bệnh nhân đã sử dụng sản phẩm.
 Chuyên gia nhắc bạn: Một sai lầm phổ biến là cho rằng nếu có thể đo đạc thì không cần Validation. Hãy nhớ: Với các quy trình đặc thù như tiệt trùng, dù bạn có lấy mẫu kiểm tra thì FDA vẫn bắt buộc phải Validation vì rủi ro cho bệnh nhân là cực kỳ lớn.
Chuyên gia nhắc bạn: Một sai lầm phổ biến là cho rằng nếu có thể đo đạc thì không cần Validation. Hãy nhớ: Với các quy trình đặc thù như tiệt trùng, dù bạn có lấy mẫu kiểm tra thì FDA vẫn bắt buộc phải Validation vì rủi ro cho bệnh nhân là cực kỳ lớn.
 Danh sách kiểm tra (Checklist) dành cho bạn:
Danh sách kiểm tra (Checklist) dành cho bạn:
- [ ] Kết quả đầu ra có thể kiểm tra 100% mà không làm hỏng sản phẩm không? (Nếu KHÔNG -> Phải Validation).
- [ ] Các sai sót có khả năng chỉ xuất hiện khi bệnh nhân đã sử dụng sản phẩm không? (Nếu CÓ -> Phải Validation).
- [ ] Quy trình có nằm trong danh mục “nhạy cảm” (Tiệt trùng, phần mềm y tế) không? (Nếu CÓ -> Bắt buộc Validation).
Ví dụ về các quy trình bắt buộc phải Validation
| Loại quy trình | Ví dụ thực tế | Tại sao không thể chỉ kiểm tra (Verification)? |
| Tiệt trùng | Khí EO, bức xạ, nhiệt ẩm | Kiểm tra độ vô trùng là “kiểm tra phá hủy”. Bạn không thể bán một sản phẩm đã bị bóc vỏ để test vi khuẩn. |
| Đúc tiêm | Vỏ máy nhựa, ống nghiệm | Các khuyết tật bên trong cấu trúc nhựa có thể gây nứt vỡ dưới áp lực khi đang sử dụng trên cơ thể. |
| Hàn túi tiệt trùng | Hàn nhiệt túi đựng thiết bị | Nếu đường hàn hở dù chỉ vài micron, vi khuẩn sẽ xâm nhập. Bạn không thể kiểm tra độ kín 100% mà không bóc túi. |
| Phần mềm (Software) | Phần mềm điều khiển máy thở | Một lỗi code (bug) có thể chỉ xuất hiện trong một điều kiện hiếm gặp khi đang cấp cứu bệnh nhân. |
| Xử lý nhiệt | Làm cứng đinh vít xương | Sự thay đổi cấu trúc kim loại bên trong không thể kiểm tra bằng mắt thường. |
3. Ba Trụ Cột IQ – OQ – PQ: Hiểu Đúng Để Làm Trúng
Để xây dựng một quy trình sản xuất vững chãi, chúng ta cần đi qua ba giai đoạn then chốt. Hãy coi đây là quá trình “Huấn luyện” cho máy móc và con người để họ luôn làm đúng.
 IQ (Installation Qualification) – Lắp đặt thiết bị
IQ (Installation Qualification) – Lắp đặt thiết bị
IQ trả lời câu hỏi: “Máy móc đã được lắp đúng spec chưa?”. Ở giai đoạn này, chúng ta đảm bảo môi trường xung quanh sẵn sàng cho thiết bị hoạt động.
- Kiểm tra nguồn điện, nguồn khí nén, nhiệt độ phòng sạch.
- Xác nhận phần mềm đã cài đặt đúng phiên bản và đã được bảo mật.
- Chuyên gia lưu ý: IQ không chỉ là cắm điện. Bạn phải chứng minh rằng máy được đặt ở vị trí an toàn, ổn định và có đầy đủ hồ sơ bảo trì định kỳ.
OQ (Operational Qualification) – Vận hành quy trình
OQ trả lời câu hỏi: “Quy trình có hoạt động ổn định tại các giới hạn không?”. Đây là lúc chúng ta xác định Cửa sổ vận hành (Window of Operation).
- Chúng ta thử nghiệm tại các “điểm giới hạn” (worst-case). Nếu máy hàn túi hoạt động từ 150°C đến 170°C, bạn phải chứng minh sản phẩm vẫn đạt chuẩn ở cả 150°C (mức thấp nhất) và 170°C (mức cao nhất).
- Mục tiêu là tìm ra giới hạn mà tại đó quy trình vẫn tạo ra sản phẩm an toàn.
PQ (Performance Qualification) – Hiệu năng thực tế
PQ trả lời câu hỏi: “Quy trình có tạo ra sản phẩm nhất quán trong dài hạn không?”. OQ cho thấy máy chạy tốt tại các điểm biên, nhưng PQ mới là lúc chứng minh tính ổn định trong điều kiện sản xuất bình thường.
- PQ phải phản ánh các thách thức thực tế: Chạy liên tục (thường là 3 lô liên tiếp), thay đổi giữa các ca làm việc, sự khác biệt giữa các kỹ thuật viên và biến động môi trường (nhiệt độ, độ ẩm).
- Chuyên gia lưu ý: Đừng nhầm lẫn giữa việc kiểm tra máy (Equipment Qualification) với xác nhận quy trình (Process Validation). PQ tập trung vào kết quả cuối cùng trên tay bệnh nhân.
Bảng tổng hợp so sánh IQ – OQ – PQ
| Tiêu chí | IQ (Lắp đặt) | OQ (Vận hành) | PQ (Hiệu năng) |
| Mục tiêu chính | Kiểm tra phần cứng, spec máy. | Xác định “Window of Operation”. | Chứng minh tính nhất quán lâu dài. |
| Câu hỏi cốt lõi | Máy đã lắp đúng chưa? | Máy chạy ổn ở mọi mức không? | Quy trình có tạo ra hàng tốt mỗi ngày không? |
| Yếu tố thử nghiệm | Tài liệu máy, nguồn năng lượng. | Thử nghiệm tại các điểm biên (Worst-case). | Nguyên liệu thật, nhiều nhân viên, biến động môi trường. |
| Khi nào làm lại? (Triggers) | Di dời máy, thay linh kiện chính. | Thay đổi thông số cài đặt (set-points). | Thay đổi nhà cung cấp nguyên liệu, có khiếu nại chất lượng. |
4. Validation Master Plan (VMP): Bản Đồ Chiến Lược
Một nhà máy MedTech có thể có hàng trăm thiết bị. Làm sao để bạn không bị lạc lối? Đó chính là vai trò của Kế hoạch tổng thể về Validation (VMP). VMP không chỉ là một tập tài liệu; nó là bản đồ chiến lược của bạn.
3 lợi ích cốt lõi của một VMP chuẩn chỉnh:
- Quản trị rủi ro: VMP giúp bạn ưu tiên quy trình nào cần Validation trước (dựa trên mức độ nguy hiểm đối với bệnh nhân).
- Tối ưu nguồn lực: Tránh việc làm lặp lại các thử nghiệm không cần thiết, giúp tiết kiệm hàng tỷ đồng tiền vật tư thử nghiệm.
- Lời cam kết với cơ quan quản lý: Khi FDA đến, VMP là bằng chứng đầu tiên cho thấy doanh nghiệp có tầm nhìn và sự kiểm soát chủ động đối với chất lượng.
5. Góc Cập Nhật: Thuật Ngữ Mới Trong Kỷ Nguyên QMSR
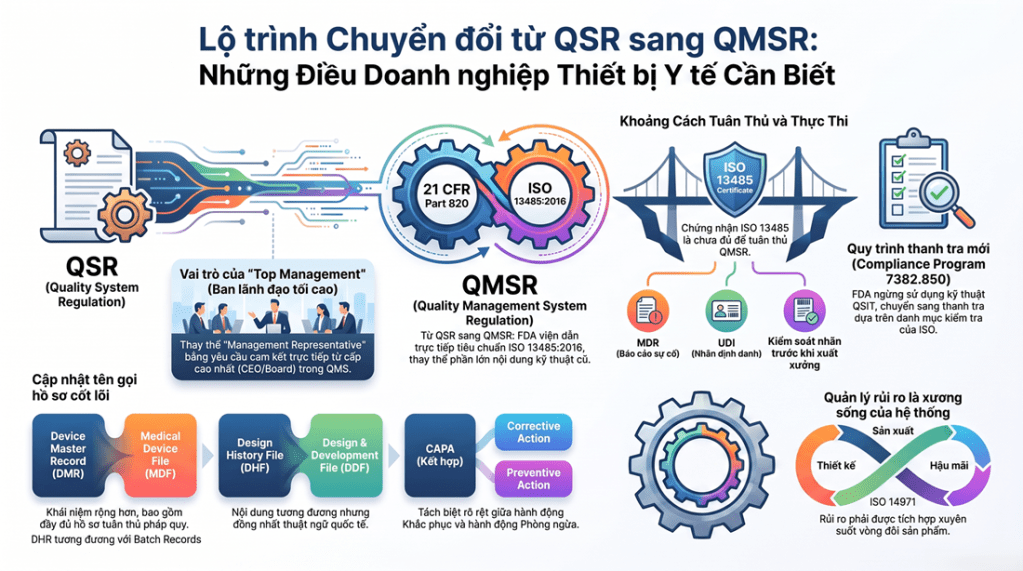
Kể từ ngày 02/02/2026, FDA chính thức áp dụng quy định QMSR (Quality Management System Regulation) để hài hòa với tiêu chuẩn quốc tế ISO 13485:2016. Đây là thay đổi lớn nhất trong hơn 25 năm qua mà mọi chuyên gia MedTech cần biết.
Bảng so sánh thuật ngữ (Từ QSR sang QMSR)
| Tên cũ (Legacy Term) | Tên mới (QMSR Term) | Bản chất thay đổi |
| DMR (Device Master Record) | MDF (Medical Device File) | Chứa mọi thông số kỹ thuật và “công thức” sản xuất. |
| DHR (Device History Record) | Batch Records (Hồ sơ lô) | Nhật ký chứng minh lô hàng đã tuân thủ đúng MDF. |
| DHF (Design History File) | DDF (Design & Development File) | Hồ sơ chứng minh quá trình thiết kế an toàn. |
 Cảnh báo đỏ từ chuyên gia:
Cảnh báo đỏ từ chuyên gia:
- ISO 13485 \neq FDA Exemption: Nhiều người tin rằng có chứng chỉ ISO 13485 là không cần lo thanh tra FDA. Sai! FDA vẫn sẽ thanh tra bạn, nhưng họ sẽ dùng kỹ thuật mới (Compliance Program 7382.850).
- Mất quyền miễn trừ: Trước đây, báo cáo Đánh giá nội bộ (Internal Audit) được FDA miễn kiểm tra. Nhưng theo QMSR 2026, FDA có quyền kiểm tra toàn bộ hồ sơ đánh giá nội bộ và xem xét biên bản họp ban lãnh đạo. Mọi thứ phải được minh bạch tuyệt đối!
6. Tổng Kết Và Thông Điệp Truyền Cảm Hứng
Process Validation không phải là một “gánh nặng hành chính” hay những chồng hồ sơ khô khan. Nó là một quá trình khoa học giúp chúng ta thấu hiểu giới hạn của máy móc và con người.
Hãy luôn ghi nhớ: Validation không chỉ là tuân thủ pháp luật, mà là lời cam kết bảo vệ sức khỏe người bệnh. Mỗi thông số bạn kiểm tra trong OQ hay mỗi lô hàng bạn chạy thử trong PQ chính là rào chắn bảo vệ sự sống cho bệnh nhân ở đâu đó ngoài kia.
 3 “Từ khóa vàng” cho sự nghiệp MedTech của bạn:
3 “Từ khóa vàng” cho sự nghiệp MedTech của bạn:
- Bằng chứng khách quan (Objective Evidence): Nếu không được ghi chép lại một cách trung thực, việc đó coi như chưa từng xảy ra.
- Tính nhất quán (Consistency): Chất lượng không phải là một sự kiện ngẫu nhiên, mà là kết quả của một quy trình được kiểm soát.
- Tư duy dựa trên rủi ro (Risk-based Thinking): Luôn bắt đầu từ câu hỏi: “Điều gì có thể gây hại cho bệnh nhân?” để thiết kế quy trình Validation.
Chúc bạn trở thành một chuyên gia chất lượng đầy bản lĩnh và tâm huyết!
Hài Hòa Hóa Tiêu Chuẩn Chất Lượng Thiết Bị Y Tế: Từ QSR (21 CFR 820) Sang QMSR & ISO 13485
1. Bối cảnh lịch sử và Bước ngoặt QMSR 2026
Trong hơn một phần tư thế kỷ, các nhà sản xuất thiết bị y tế muốn chinh phục thị trường Hoa Kỳ đều phải tuân thủ nghiêm ngặt 21 CFR Part 820, hay còn gọi là QSR (Quality System Regulation). Tuy nhiên, một kỷ nguyên mới đã bắt đầu. Vào ngày 02/02/2024, FDA đã ban hành quy định cuối cùng nhằm sửa đổi Part 820, chính thức đổi tên thành QMSR (Quality Management System Regulation).
Sự thay đổi này không chỉ là một đợt cập nhật văn bản thông thường; đó là một cuộc cách mạng về pháp lý. Bằng cách viện dẫn (incorporation by reference) tiêu chuẩn quốc tế ISO 13485:2016, FDA đã phá bỏ bức tường ngăn cách giữa quy định nội địa Mỹ và tiêu chuẩn toàn cầu. Doanh nghiệp cần đặc biệt lưu ý cột mốc: 02/02/2026. Đây là thời hạn cuối cùng và duy nhất.
“Đây là sự thay đổi lớn nhất trong hơn 25 năm qua của FDA. Việc chuyển dịch từ cấu trúc QSR sang QMSR đánh dấu bước ngoặt từ một danh mục kiểm tra (checklist) cứng nhắc, đặc thù kiểu Mỹ sang một khung quản lý quy trình (process-oriented) đồng nhất với thế giới.”
Sự thay đổi này đồng nghĩa với việc QMSR giờ đây là “luật của quốc gia” (law of the land) đối với bất kỳ thiết bị y tế nào lưu hành tại Mỹ. Nó yêu cầu một sự chuyển dịch tư duy: từ việc chỉ “đối phó” với các quy định riêng biệt sang việc vận hành một hệ thống chất lượng tích hợp toàn cầu.
2. Lý do FDA chuyển đổi từ QSR sang QMSR (Tại sao lại là lúc này?)
FDA không thực hiện sự thay đổi này vì sự tùy hứng. Đây là kết quả của nỗ lực giảm thiểu rào cản thương mại và nâng cao hiệu quả giám sát dựa trên 03 lý do cốt lõi:
- Giảm bớt gánh nặng hành chính và chi phí tuân thủ: Trước đây, các doanh nghiệp xuất khẩu thường phải duy trì hai bộ Sổ tay Chất lượng (Quality Manual) với hệ thống đánh số và thuật ngữ khác nhau để làm hài lòng cả FDA và các tổ chức chứng nhận ISO.
- Giá trị thực tế: Doanh nghiệp không còn phải lãng phí nguồn lực để vận hành hai hệ thống song song, giúp tối ưu hóa nhân sự chất lượng vào việc cải tiến sản phẩm thực hại thay vì quản trị hồ sơ dư thừa.
- Hài hòa hóa quốc tế và đồng bộ hóa giám sát: Việc sử dụng chung “ngôn ngữ” ISO 13485 tạo điều kiện cho các chương trình như MDSAP (Chương trình Đơn kiểm Thiết bị Y tế) phát triển mạnh mẽ hơn.
- Giá trị thực tế: Tạo ra một môi trường pháp lý minh bạch, nơi kết quả kiểm tra có thể được chia sẻ và công nhận rộng rãi hơn giữa các quốc gia (như EU, Canada, Nhật Bản).
- Thúc đẩy tiếp cận thiết bị an toàn nhanh hơn: Bằng cách loại bỏ những khác biệt không cần thiết trong quy định, FDA giúp giảm bớt “rào cản kép” cho các nhà sản xuất.
- Giá trị thực tế: Đặc biệt với các doanh nghiệp Việt Nam, việc này giúp rút ngắn đáng kể thời gian đưa sản phẩm vào thị trường Mỹ (Time-to-market) khi hệ thống hiện tại đã đáp ứng sẵn tiêu chuẩn quốc tế.
Sự chuyển dịch này không chỉ là thay đổi về cấu trúc mà còn là sự thay đổi sâu sắc về ngôn ngữ kỹ thuật mà chúng ta sẽ đối chiếu ngay sau đây.
3. Bảng đối chiếu thuật ngữ: QSR cũ vs. QMSR (ISO 13485)
Là một kiểm sát viên, tôi yêu cầu các bạn phải chuẩn bị sẵn một “ma trận đối chiếu” (mapping matrix) trong hệ thống tài liệu của mình. Lưu ý quan trọng: Định nghĩa của FDA (Part 820.3) vẫn giữ quyền ưu tiên cao nhất so với định nghĩa ISO trong trường hợp có sự xung đột.
| Thuật ngữ QSR cũ (FDA) | Thuật ngữ QMSR/ISO mới | Ghi chú thay đổi quan trọng |
| DHF (Design History File) | Design and Development File (DDF) | Tên gọi thay đổi nhưng bản chất lưu trữ hồ sơ thiết kế vẫn duy trì nghiêm ngặt. |
| DMR (Device Master Record) | Medical Device File (MDF) | MDF có phạm vi rộng hơn, bao gồm cả mô tả thiết bị, mục đích sử dụng và các hồ sơ kỹ thuật hỗ trợ. |
| DHR (Device History Record) | Batch Record / Medical Device Record | Hồ sơ bằng chứng về việc sản xuất cho từng lô sản phẩm cụ thể. |
| Establish (Thiết lập) | Document (Văn bản hóa) | Đây là thay đổi ngữ nghĩa then chốt. ISO dùng “Document” để bao hàm cả việc định nghĩa, triển khai và duy trì. |
| Management Representative | Top Management | FDA hiện yêu cầu sự cam kết trực tiếp từ lãnh đạo cao nhất (CEO/Board) thay vì chỉ ủy quyền cho một cá nhân. |
| Quality System (QS) | Quality Management System (QMS) | Nhấn mạnh vào tính quản trị hệ thống thay vì chỉ là các bước kỹ thuật đơn thuần. |
Dù tên gọi có thể thay đổi, nhưng hãy nhớ rằng bản chất cốt lõi của việc lưu trữ bằng chứng khách quan vẫn là yêu cầu tối thượng trong mọi cuộc thanh tra.
4. Mối quan hệ giữa Chứng nhận ISO 13485 và Tuân thủ FDA
Đây là nơi mà nhiều doanh nghiệp Việt Nam thường mắc sai lầm chết người. Với tư cách là chuyên gia cố vấn, tôi phải đưa ra cảnh báo sau:
CẢNH BÁO QUAN TRỌNG
Chứng nhận ISO 13485 KHÔNG đồng nghĩa với việc mặc nhiên tuân thủ QMSR.
- Tài sản thương mại vs. Nghĩa vụ pháp lý: Chứng chỉ ISO 13485 là một “tài sản thương mại” tự nguyện, trong khi tuân thủ QMSR là một “mệnh lệnh pháp lý” bắt buộc theo Đạo luật FD&C.
- FDA không cấp chứng chỉ: FDA không cấp giấy chứng nhận như các tổ chức Bureau Veritas hay BSI. Họ thực hiện thanh tra đột xuất hoặc định kỳ để thực thi pháp luật.
- Sự khác biệt về mục tiêu: Một chuyên gia đánh giá ISO tập trung vào sự phù hợp (conformance), trong khi thanh tra viên FDA tập trung vào sự tuân thủ luật pháp (compliance). Một thiết bị không tuân thủ QMSR sẽ bị coi là “adulterated” (kém phẩm chất) và bị từ chối nhập khẩu ngay lập tức.
5. Các yêu cầu bổ sung đặc thù của FDA (Phần nằm ngoài ISO 13485)
FDA không chấp nhận ISO 13485 một cách nguyên bản 100%. Họ đã “cấy” thêm các yêu cầu đặc thù của luật pháp Hoa Kỳ tại các mục §820.10, §820.35, và §820.45:
- Báo cáo sửa chữa và thu hồi (Advisory Notices – Part 806): ISO yêu cầu “Phản hồi”, nhưng FDA yêu cầu quy trình báo cáo pháp lý nghiêm ngặt về các hành động khắc phục và thu hồi sản phẩm khỏi thị trường.
- Truy xuất nguồn gốc cho thiết bị duy trì sự sống (§820.10 / Part 821): Đây là yêu cầu khắt khe hơn ISO. FDA bắt buộc các thiết bị cấy ghép hoặc hỗ trợ sự sống phải có hệ thống truy xuất đến tận người dùng cuối.
- Nhận dạng thiết bị duy nhất (UDI): FDA bắt buộc ghi nhận mã UDI trong mọi giai đoạn của vòng đời sản phẩm, một yêu cầu mà ISO chỉ đề cập chung chung.
- Kiểm soát ghi nhãn và bao bì (§820.45): FDA giữ lại yêu cầu riêng về việc kiểm tra độ chính xác của nhãn mác trước khi xuất xưởng để ngăn ngừa nhầm lẫn sản phẩm – nguyên nhân hàng đầu của các vụ thu hồi.
- Hồ sơ khiếu nại phải đủ 07 trường thông tin (§820.35): Bao gồm: tên thiết bị, ngày nhận, mã UDI, thông tin liên lạc người khiếu nại, chi tiết khiếu nại, hành động khắc phục và phản hồi.
- Hồ sơ dịch vụ (Servicing) phải đủ 06 trường thông tin: Bao gồm tên thiết bị, mã nhận dạng (UDI/Serial), ngày bảo trì, cá nhân thực hiện, nội dung dịch vụ và dữ liệu kiểm tra/thử nghiệm.
6. Thay đổi trong công tác Thanh tra và Kiểm soát rủi ro
Cách thức FDA “gõ cửa” doanh nghiệp sẽ thay đổi hoàn toàn sau tháng 02/2026. Kỹ thuật thanh tra QSIT cũ sẽ bị khai tử, thay thế bằng Chương trình Tuân thủ 7382.850 mới, được thiết kế theo cấu trúc điều khoản của ISO 13485.
Phá bỏ “Vùng cấm hồ sơ”: Đây là thay đổi chấn động nhất. Trước đây, hồ sơ đánh giá nội bộ (internal audits), đánh giá nhà cung cấp và xem xét của lãnh đạo (management reviews) được hưởng quyền miễn trừ kiểm tra của FDA. Hiện nay, các hồ sơ này đã trở thành “cuốn sách mở” đối với thanh tra viên. Nếu nội bộ công ty phát hiện lỗi mà không lập tức đưa vào quy trình CAPA, FDA sẽ coi đó là sự che giấu vi phạm và đưa ra các biện pháp chế tài nặng nề.
Quản lý rủi ro (ISO 14971) không còn là lựa chọn: Theo QMSR, quản lý rủi ro không còn chỉ nằm gọn trong hồ sơ thiết kế. Nó phải được tích hợp vào tất cả các quy trình từ Mua hàng, Sản xuất đến Dịch vụ hậu mãi. Nếu bạn không thể chứng minh việc đánh giá rủi ro khi thay đổi nhà cung cấp linh kiện, hệ thống của bạn sẽ bị đánh giá là không đạt.
7. Tầm quan trọng của sự hài hòa hóa toàn cầu
Việc chuyển đổi sang QMSR là cơ hội vàng để các doanh nghiệp Việt Nam chuẩn hóa năng lực cạnh tranh. Để chuẩn bị, các bạn cần thực hiện 03 bước hành động ngay lập tức:
- Phân tích khoảng cách (Gap Analysis) ngay bây giờ: Đừng đợi đến năm 2026. Hãy rà soát xem các hồ sơ DHF, DMR hiện tại có thiếu hụt các trường thông tin đặc thù mà FDA yêu cầu hay không.
- Đồng bộ hóa thuật ngữ: Cập nhật lại Sổ tay Chất lượng và các SOPs theo ngôn ngữ mới (MDF thay cho DMR, DDF thay cho DHF) để đảm bảo “nói cùng ngôn ngữ” với thanh tra viên.
- Tích hợp quản lý rủi ro sâu rộng: Đưa ISO 14971 vào mọi khía cạnh vận hành của QMS như một yêu cầu pháp lý bắt buộc.
Kết luận: Sự hài hòa hóa giữa FDA và ISO không chỉ là nỗ lực cắt giảm thủ tục. Đó là thông điệp rằng chất lượng không còn là việc “vượt qua một cuộc kiểm tra”, mà là nền tảng của sự cạnh tranh toàn cầu. Hãy xây dựng một Văn hóa Chất lượng thực thụ, nơi sự an toàn của bệnh nhân là kim chỉ nam, chứ không chỉ là những tệp hồ sơ nằm chờ trên kệ sách. Thời hạn 02/02/2026 không có giai đoạn chuyển tiếp (no phase-in period) – hãy hành động trước khi quá muộn.
Liên hệ tải văn bản tài liệu liên quan pdf song ngữ Anh Việt: chỉ từ 50$
Chúng tôi cung cấp tài liệu tiêu chuẩn bản gốc và bản dịch
• Bản gốc tiếng anh của Tiêu Chuẩn sơ đồ tóm tắt và hình ảnh minh họa rõ ràng. Để quý khách thuận tiện đối chiếu theo từng điều khoản, dễ tra cứu và lập hồ sơ.
• Bản dịch tiếng Việt chuẩn chuyên ngành, dùng được ngay cho công việc.
• Ghi chú diễn giải và hướng dẫn áp dụng từ chuyên gia thực chiến.
• Giao nhanh dưới định dạng PDF/Word thuận tiện chỉnh sửa, lưu trữ.
Phạm vi tiêu chuẩn
Hệ thống quản lý ISO (9001, 14001, 45001, 37001…..); chứng nhận bền vững (ISCC, FSC, VFCS/PEFC….); dệt may và vật liệu tái chế (GRS, OCS, OEKO-TEX….); trách nhiệm xã hội và ESG (SMETA, EcoVadis, Fairtrade….) cùng nhiều tiêu chuẩn ngành khác theo yêu cầu.
Vì sao chọn chúng tôi
Tài liệu được biên dịch bởi đội ngũ trực tiếp tư vấn và đánh giá chứng nhận, nên bản dịch không chỉ đúng ngôn ngữ mà còn đúng tinh thần áp dụng thực tế — giúp doanh nghiệp tiết kiệm thời gian và hạn chế sai sót khi xây dựng hồ sơ.
Chi phí
Chỉ từ 50 USD/tài liệu, tùy độ dài và độ phức tạp. Quý Doanh nghiệp vui lòng gửi tên tiêu chuẩn để nhận báo giá chính xác miễn phí.
Cách đăng ký
1. Liên hệ và cho biết tên tiêu chuẩn cần dịch.
2. Nhận báo giá và thời gian giao tài liệu.
3. Thanh toán và nhận tài liệu qua email.
Thanh toán
Chuyển khoản qua mã QR ngân hàng (gửi kèm khi báo giá), hoặc qua PayPal: ducluongservices@gmail.com.
Rất mong được đồng hành cùng Quý Doanh nghiệp trên hành trình chứng nhận.
Trân trọng,
THÔNG TIN LIÊN HỆ
Hotline: +84 933 096 426 – +84 868 591 260
Email: info@staunchlyservices.com.vn | info@iscglobal.asia | van.pham@iscglobal.asia | ducluongservices@gmail.com





